
Wilson Disease, also known as Wilson’s Disease or hepatolenticular degeneration, was first described by Samuel Wilson in 1912 in the context of neurodegenerative symptoms and liver damage. In his manuscript, Wilson called the disease “progressive lenticular degeneration,” especially concerned with the presentation of neurodegenerative symptoms [57]. In his description of the disease, Wilson detailed various neurological symptoms and noted cirrhotic liver in post mortem analyses of patients.Wilson also noticed a characteristic and uncontrollable “spastic smile,” elbows, hands, and feet bent into fixed positions, and tremors. Some patients were unable to swallow or had extreme difficulty swallowing, and the majority of patients lacked complete motor control. Shortly before death, patients were extremely emaciated. Despite cirrhotic liver in deceased patients, Wilson did not cite other symptoms of liver damage in several cases [57]. Wilson also recognized that the disease was familial, and surmised that the disease was of autosomal recessive inheritance.
Prior to Wilson’s description of progressive lenticular degeneration, Kayser and Fleischer described a brown ring in the cornea surrounding the iris in 1902 and 1912, respectively, which would later become pathognomonic of Wilson Disease [20]. In 1933, Wilson observed the ring in two patients who had liver cirrhosis, and citing the exceedingly rare nature of both conditions, believed that they must be linked in some way [58]. One of Wilson’s colleagues, T. Harrison Butler, believed that the condition was driven by significant accumulation of copper in the liver, potentially accompanied by manganese poisoning. It was shown in 1936 that the Kayser-Fleischer ring was composed of deposited copper, lending evidence to the idea that Wilson Disease was driven by a disturbance in copper metabolism [20].

A photograph of a patient with the Kayser-Fleischer ring present around the iris of the eye. In this picture, you can see a distinct darkness surrounding the iris and a slight browning within the ring. Image from Harry and Tripathi [20].
When it was established that Wilson Disease was driven by the accumulation of copper, chelation therapies were developed. 2,3-dimercaptopropanol (BAL), which functions as a chelating agent, was first used to treat Wilson Disease in 1951 and many patients saw significant improvements following treatment [9].
In 1952, it was first shown Wilson Disease patients have a deficiency in serum ceruloplasmin, a copper carrying protein. It had been shown previously that serum copper concentrations were abnormal in cases of Wilson Disease, and Scheinberg and Gitlin looked into the role of ceruloplasmin in this abnormality. Because the majority of copper in the blood is bound to ceruloplasmin, an abnormal serum copper concentration could naturally be driven by a disturbance in ceruloplasmin. In their research, Scheinberg and Gitlin determined that ceruloplasmin levels were significantly lower in Wilson Disease patients, though it was still unclear how this depreciation of serum ceruloplasmin was connected to the disease [45]. A general belief was that Wilson Disease was driven by a disruption in ceruloplasmin synthesis, and that the lack of ceruloplasmin promoted the accumulation of copper that was seen in all patients [46]. Ceruloplasmin is essential for effective copper circulation to tissues from the liver; the implications of reduced serum ceruloplasmin will be expanded upon in the following pages.
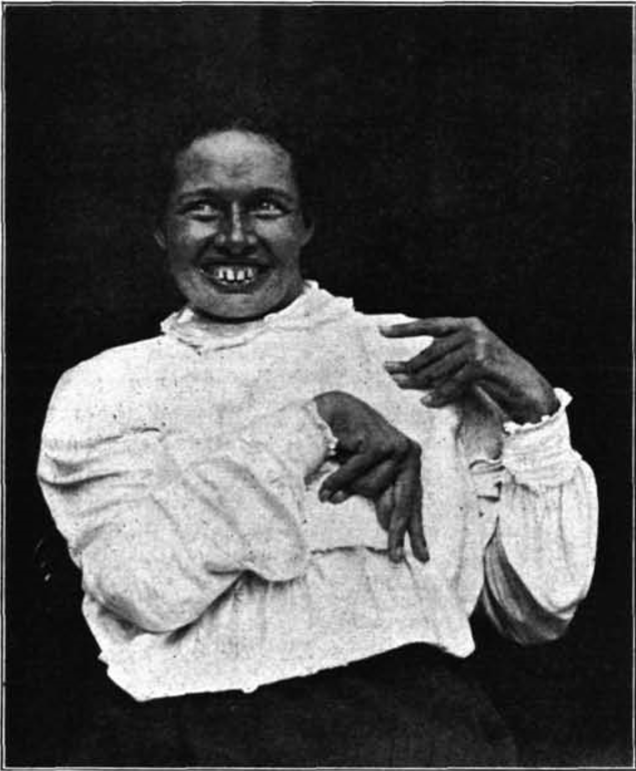
A photograph of one of Samuel Wilson’s patients expressing neurological symptoms. Of note is the uncontrolled spastic smile and forcibly bent elbows and hands, all of which dominated the physical state of the patient. Wilson also describes an occasional lack of control in eye movements and extreme difficulty in swallowing food [57].
Ceruloplasmin deficiency was the most consistent manifestation of Wilson Disease, which supported the belief that this deficiency was the root cause of the condition. In 1959, however, it was shown that ceruloplasmin levels were normal and maintained over the course of several years in two brothers with clear Wilson Disease symptoms, including the Kayser-Fleischer ring [44]. Despite having typical serum ceruloplasmin levels, one of the brothers incorporated copper into ceruloplasmin at a markedly slower rate. With this work, they showed that the disease is not driven by a failure to effectively synthesize ceruloplasmin, which challenged a popular belief concerning Wilson Disease pathogenesis. This discovery was important to future understanding of the disease, as it was later found that the characteristic ceruloplasmin deficiency was not a cause of the disease but rather the result of a separate genetic mutation that failed to utilize ceruloplasmin in an essential pathway.
Sternlieb et al. determined in 1973 that copper concentration in bile was significantly decreased in Wilson Disease patients, a major discovery in understanding the cause of Wilson Disease symptoms. More importantly, they find that lysosomal copper activity is directly linked to biliary copper activity, thus identifying a connection between lysosomal activity and biliary copper secretion [51]. As discussed in page 1, lysosomes serve as an intermediate in the copper excretion pathway and interact with ATP7B to eliminate excess intracellular copper. Additionally, copper contained in lysosomes is nontoxic, whereas cytoplasmic copper is cytotoxic, and reduced biliary excretion of copper increases the concentration of copper in the cytoplasm of hepatic cells, as biliary excretion is the primary mode of copper excretion in mammals [12, 23].
A diagram of copper import and export in hepatic cells from Terada et al shows the basic movement of copper with (A) and without ATP7B (B). The importance of the lysosome in copper excretion is of note, as well as the inability to load ceruloplasmin (CPN), which leads to its eventual degradation [53].
Despite massive advances in the understanding of Wilson Disease, the basic molecular cause of the disease remained unknown. As technology advanced and more gene-evaluating techniques were developed, the potential to find the molecular culprit increased. In 1990, Frydman expanded on his previous work and mapped the Wilson Disease gene to chromosome 13 with high confidence [13]. Though it was not unequivocally proven in this work, Frydman showed that Wilson Disease was more than likely caused by a single gene as opposed to a cluster of genes responsible for copper metabolism.
Just a few years later, the molecular culprit was found. In 1993, Tanzi et al. showed that the Wilson Disease gene was ATP7B, a copper transporter ATPase with homology to ATP7A, another copper transporter ATPase that has also been found to cause disruptions in copper metabolism [52]. It was this work that finally identified the molecular cause of Wilson Disease, and significantly advanced research in the field. After determining the gene responsible, work shifted to focus primarily on what mutations drove the disease and why these mutations were so disruptive of copper metabolism in hepatic cells. In the following years, several novel mutations were found to cause Wilson Disease [27].
Wilson Disease is now categorized as a rare condition, with an incidence rate between 1/30,000 and 1/100,000 with an estimated carrier incidence of 1/90, though the proportion of affected individuals varies by location and across populations. In some areas, 1/10,000 births may result in a child born with Wilson Disease.